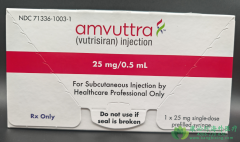
遗传性转甲状腺素蛋白淀粉样变性(ATTRv)患者长期面临“诊断难、治疗无”的困境,随着神经与心脏功能的不可逆衰退,疾病成为吞噬生命的“沉默杀手”。武特里西兰,这一基于RNA干扰技术的创新疗法,通过精准沉默致病基因的表达,彻底改写了遗传性神经病的治疗格局,为患者提供了延缓疾病恶化的关键武器,开启了治疗新纪元。

ATTRv的致命性源于转甲状腺素蛋白(TTR)的异常沉积,导致周围神经与心脏组织逐渐损毁。武特里西兰的治疗逻辑直击这一病理核心:通过皮下注射的siRNA分子,精准识别并降解TTRmRNA,从基因层面抑制TTR蛋白合成。与传统疗法(如缓解症状或稳定蛋白构象)不同,其创新性在于直接“关闭”致病基因的“生产开关”,从源头阻止淀粉样沉积的形成,尤其在遗传性多发性神经病(ATTR-PN)中展现出强大的疾病修饰能力,为患者保留了更多功能与生存时间。
该药物专用于成人ATTR-PN的治疗,无论突变型或野生型TTR相关疾病。每三个月一次的皮下注射方案,极大简化了治疗流程,避免了每日服药或频繁医疗干预,显著提升患者生活质量。治疗期间需关注注射部位反应(如短暂红肿)、监测潜在过敏反应,尤其首次注射时需严密观察。长期需定期评估神经病变进展(如通过神经评分系统)和心脏功能,警惕肝脏酶学指标异常,确保治疗安全。
临床试验数据彰显武特里西兰的突破性价值。APOLLO研究证实,治疗18个月后,患者神经病变评分(mNIS+7)年恶化率较安慰剂组降低超80%,患者行走能力、日常生活活动能力显著稳定,且因疾病进展导致的住院风险大幅下降。对比历史对照数据,其延缓疾病进展的幅度远超现有疗法,部分患者病情甚至出现轻微改善。这一成果彻底颠覆了“ATTRv不可逆转”的认知,为患者带来了实质性的生存获益。
患者王先生的案例极具代表性。他确诊ATTR-PN后,双腿麻木逐渐加重,最终无法独立行走,需依赖轮椅。使用武特里西兰两年后,神经病变恶化速度显著放缓,疼痛减轻,手部力量部分恢复,能够重新进行简单的户外活动。此类案例凸显了武特里西兰在改写疾病轨迹与重塑生活希望中的关键作用。
在药物对比层面,武特里西兰优势鲜明。相较于口服小分子药物(如tafamidis),其直接靶向mRNA的疗法在作用机制上更根本,且长效注射避免了每日用药的依从性挑战;对比传统支持疗法,其疾病修饰作用实现了从“治标”到“治本”的跨越。尽管tafamidis在稳定TTR蛋白构象方面有效,武特里西兰为疾病进展更快或对口服药物不耐受的患者提供了救命选择,尤其在延缓神经病变恶化速度与长期安全性方面表现卓越。
尽管武特里西兰已取得重大突破,其应用仍需强调多学科协作,早期精准诊断与基因分型是关键。未来,其在心脏型ATTRv中的应用潜力、长期安全性数据积累及更便捷的给药方式优化是发展方向。随着更多临床数据的验证,其治疗地位将更加稳固。
作为改写遗传性神经病治疗格局的创新突破,武特里西兰不仅为患者提供了前所未有的疾病控制手段,更重新定义了罕见病治疗的可能性。它深刻诠释了基因技术如何将“无药可医”转化为“精准干预”,为深陷ATTRv困境的患者开启了生存与尊严的新篇章。如有需要,请咨询康必行海外医疗医学顾问:4006-130-650或扫码添加下方微信,我们将竭诚为您服务!
更多药品详情请访问 武特里西兰 https://www.kangbixing.com/drug/wtlxl/
 添加康必行顾问,想问就问
添加康必行顾问,想问就问