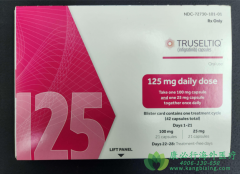
在晚期胆管癌的治疗版图中,FGFR2融合突变曾是一道难以跨越的障碍。这类突变占胆管癌患者的百分之五至十,因FGFR激酶异常活化,持续驱动MAPK、PI3K-AKT等增殖信号,导致肿瘤快速进展。传统化疗(如吉西他滨联合顺铂)客观缓解率仅约百分之十五,中位无进展生存期短于六个月,患者长期面临疗效有限与毒性负担的双重压力。英菲格拉替尼(Infigratinib,商品名Truseltiq)的出现,作为高选择性FGFR1-3抑制剂,通过精准阻断FGFR异常信号,为FGFR融合突变实体瘤(尤其胆管癌)提供了口服靶向调控的新可能,其核心价值在于以结构适配实现高效干预,改写无药可用的治疗困局。
FGFR融合突变的病理本质与干预挑战
成纤维细胞生长因子受体(FGFR)家族包括FGFR1-4,其正常功能是通过配体结合激活下游信号,调控细胞增殖与分化。在胆管癌、尿路上皮癌等肿瘤中,FGFR2(偶见FGFR1/3)基因与其他基因(如BICC1、AHCYL1)发生融合,导致激酶结构域持续暴露,无需配体即可自我激活,驱动肿瘤生长。这种突变在胆管癌中占比约百分之五至十,是明确的驱动因素,但因FGFR与其他激酶结构相似,早期泛靶点药物难以实现选择性抑制,脱靶毒性限制了临床应用。

英菲格拉替尼的分子设计与作用机制
英菲格拉替尼的核心优势在于对FGFR1-3激酶的高选择性阻断。其分子结构经优化,能精准嵌入FGFR激酶域的ATP结合口袋,通过氢键与疏水作用稳定结合,干扰激酶自磷酸化及下游信号传导。与早期FGFR抑制剂相比,其对FGFR1-3的选择性倍数超百倍(对VEGFR2、PDGFR等激酶抑制活性低千倍以上),这种精准匹配减少了对正常细胞(如肝细胞、血管内皮)的干扰。作用机制上,它通过阻断FGFR融合蛋白的异常激活,下调MAPK、PI3K-AKT通路活性,抑制肿瘤细胞增殖并诱导凋亡,同时可能增强肿瘤免疫原性,为联合治疗提供理论基础。
临床疗效在FGFR融合胆管癌中的验证
关键II期临床试验(NCT03773302)为英菲格拉替尼的获批提供了核心证据。该研究纳入108例经治的FGFR2融合突变晚期胆管癌患者,给予英菲格拉替尼每日一次口服125毫克(用三周停一周),结果显示客观缓解率达百分之二十六,疾病控制率百分之八十三,中位无进展生存期十一点六个月,中位总生存期三十一点一个月。亚组分析表明,无论融合伴侣基因(如BICC1、AHCYL1)或既往治疗线数(一至三线),均观察到一致的缓解趋势;基线合并肝转移者(占百分之七十二)同样获益,证实药物对全身病灶的活性。这些数据支持其作为FGFR2融合胆管癌的二线标准治疗。
临床应用需以规范化分子检测为前提
英菲格拉替尼的获批适应症为既往接受过治疗的FGFR2融合突变局部晚期或转移性胆管癌成人患者。用药前必须通过肿瘤组织或液体活检(二代测序)明确FGFR2融合(需排除FGFR1/3突变或扩增),推荐使用经认证的检测panel。标准剂量为每日一次口服125毫克,连续服用三周后停药一周(四星期为一周期),疗程持续至疾病进展或不可耐受毒性。疗效评估依赖每六至八周的影像学检查(RECIST标准),同时监测循环肿瘤DNA中FGFR融合丰度变化――丰度下降大于等于百分之五十常预示深度缓解。
安全性管理聚焦FGFR抑制的特异性反应
英菲格拉替尼最常见的不良反应为高磷血症(发生率百分之七十三,与FGFR抑制减少肾脏磷排泄相关),多数为轻中度(血磷水平小于七毫克/分升),可通过低磷饮食或短期停药缓解;眼毒性(发生率百分之五十九,包括干眼、角膜混浊),需定期眼科检查(每三至六个月裂隙灯评估),出现大于等于二级病变时暂停用药并给予人工泪液;胃肠道反应(恶心百分之四十五、腹泻百分之三十八),多为轻中度,对症处理可缓解。少见但需警惕的事件包括手足综合征(发生率百分之十二)、脱发(百分之二十一),总体因不良反应停药率约百分之十五。
英菲格拉替尼的意义与未来方向
英菲格拉替尼的上市标志着FGFR融合突变肿瘤治疗进入精准调控时代。作为首个获批用于胆管癌的FGFR抑制剂,它不仅将FGFR2融合患者的客观缓解率提升至百分之二十六(远超化疗的百分之十五),更验证了高选择性激酶抑制在实体瘤中的可行性――通过解析突变蛋白的独特激活模式,实现高效低毒干预。其临床定位提示,未来胆管癌分子分型需更关注FGFR等罕见驱动基因,探索英菲格拉替尼与免疫检查点抑制剂(如PD-1单抗)的联合(利用信号阻断增强免疫应答)、在更早期患者中的一线应用,以及与FGFR抗体偶联药物的序贯策略,将进一步拓展其价值。对患者而言,英菲格拉替尼带来的是从化疗依赖到靶向调控的转变,为FGFR融合突变这一小众亚群提供了掌控预后的新可能。如有需要,请咨询康必行海外医疗医学顾问:4006-130-650或扫码添加下方微信,我们将竭诚为您服务!
 添加康必行顾问,想问就问
添加康必行顾问,想问就问